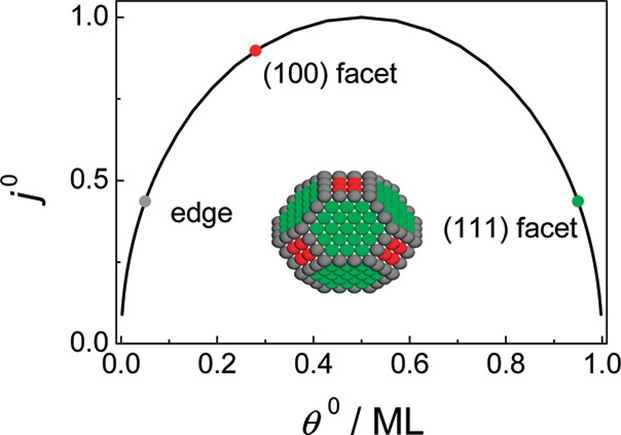
Microkinetic modeling and density functional theory (DFT) calculations are combined to understand the surface structure and nanoparticle size effects of Pt on the kinetics of hydrogen electrode reactions (HERs). The microkinetic modeling leads to a mechanism-free volcano relation between the exchange current density of HERs (j0) and the surface coverage of the reactive H adatoms at the equilibrium potential (θ0), making the activity trend of various catalytic surfaces for HERs predictable with θ0. It is shown that catalytic surfaces with θ0 values closer to 0.5 monolayer will have higher j0. A DFT calculation scheme is developed to determine the nature of the reactive H atoms and the corresponding θ0 values. The calculated results on Pt single crystal electrodes predict that j0 follows a trend that Pt(110) ≈ Pt(100) > Pt(111), whereas for Pt nanoparticles the j0 follows a trend that (100) facets > (111) facets ≈ edge rows, which in turn suggests a decrease of j0 with the decreasing particle size of Pt. It is shown that, although the individual edge atom rows on Pt fcc nanoparticles are similar in structures to the top atom rows on the Pt(110) surface, the catalytic properties of the nanoparticle edges are not simply equivalent to the extended (110) surfaces since some of the reactive sites for a reaction on extended (110) surfaces (e.g., the long-bridge sites) are absent at nanoparticle edges. The results presented here not only throw new insights into the long-standing problem about the Pt surface structure and particle size effects in hydrogen electrocatalysis but also provide a general theoretical scheme to identify the most active catalytic surfaces for HERs. More importantly, we demonstrate that not only the thermodynamic data like the adsorption energy but also a detailed nature of the reactive sites and their coverage are very important for the proper prediction of the activity trend of catalytic surfaces.
https://pubs.acs.org/doi/10.1021/jp207015w


 LINK
LINK