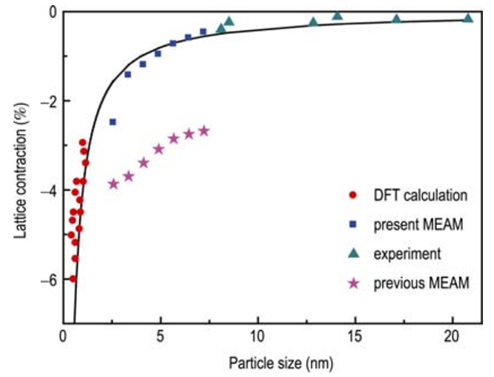
A density functional theory (DFT)-calculation scheme for constructing the modified embedded atom method (MEAM) potentials for face-centered cubic (fcc) metals is presented. The input quantities are carefully selected and a more reliable DFT approach for surface energy determination is introduced in the parameterization scheme, enabling MEAM to precisely predict the surface and nanoscale properties of metallic materials. Molecular dynamics simulations on Pt and Au crystals show that the parameterization employed leads to significantly improved accuracy of MEAM in calculating the surface and nanoscale properties, with the results agreeing well with both DFT calculations and experimental observations. The present study implies that rational DFT parameterization of MEAM may lead to a theoretical tool to bridge the gap between nanoscale theoretical simulations and DFT calculations.
https://link.springer.com/article/10.1007/s11426-010-0069-0



 LINK
LINK