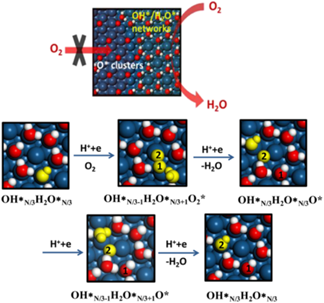
This work aims to reveal the origin of the large onset overpotential and sluggish kinetics of the oxygen reduction reaction (ORR) on Pt, a standing problem that hinders the progress of fuel cell technology. By investigating various possible reaction steps and pathways through detailed DFT calculations on Pt(111) surface covered by rationalized phase structures of oxygenated adsorbates, we show that the ORR overpotential and Tafel kinetics originate from the potential-dependent formation of a site-blocking spectator phase, √3 × √3-structured oxygen adatoms (O*), which coexists with a relatively weak blocking phase, (√3 × √3)R30°-patterned adsorption network of hydroxyl group (OH*) and water molecule (H2O*) at ORR relevant potentials. The OH*/H2O* phase provides sites for ORR to proceed through a dissociative pathway consisting of four proton/electron transfer (PET) steps. The first step, PET-coupled O2 adsorption, is identified as the activity-determining step. Different from the usual beliefs, we found the O2 and O* do not directly accept proton during the reduction steps; rather, OH* and H2O* act as PET mediators to facilitate the O2 adsorption and dissociation and the O* reduction. These findings unveil the distinctly multiple roles of various oxygenated adsorbates as intermediates, spectators, and PET mediators in ORR. The implications of these findings on designing Pt-based catalysts are discussed. It is concluded that the binding strength of O* impacts the ORR activity of Pt-based surface predominantly by modulating the number of the available active sites, rather than the activation barriers for the rate-determining step.
https://doi.org/10.1021/acs.jpcc.7b01082


 LINK
LINK