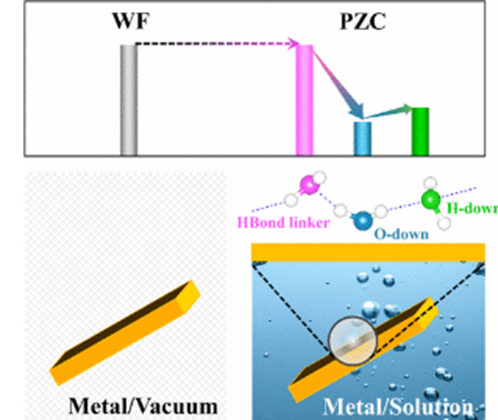
Metal–water interactions are investigated using ab initio molecular dynamics simulations performed on water-interfaced Pt(111) and Au(111) as model systems, aiming at understanding the mechanism of interface water molecules to regulate the potential of zero charge (PZC) of metal electrodes in aqueous solutions. Several metal–water interactions are distinguished, and their effects on the metal work function (WF) are quantified through carefully correlating the interfacial atomic and electronic structures. The first layer of interface water molecules possesses an O-down configuration and significantly lowers the metal WF by increasing the near-surface electron density through Pauli repulsion, coordination bonding, and subordinate dipole orientation. In contrast, the H-down-configured water molecules in the second solvation layer increase the metal WF due to the metal–hydrogen bonding interaction and dipole orientation. Involved in the second layer are also water molecules that have no preferred orientation and merely act as hydrogen bond linkers. They negligibly affect the electronic structure of metal electrodes. Introducing chemisorbed hydrogen (Had) with varying coverages modulates the metal–water interactions, resulting in a nonmonotonic variation of the metal WF. The atomic insights obtained not only help to enunciate the long-standing puzzle of a significant decrease in the PZC of metal electrodes by solvation but also add to our understanding of the behaviors of metal–solution interfaces, for examples, the potential- and adsorbate-dependent interfacial capacitance.
https://doi.org/10.1021/acs.jpcc.0c11089


 LINK
LINK