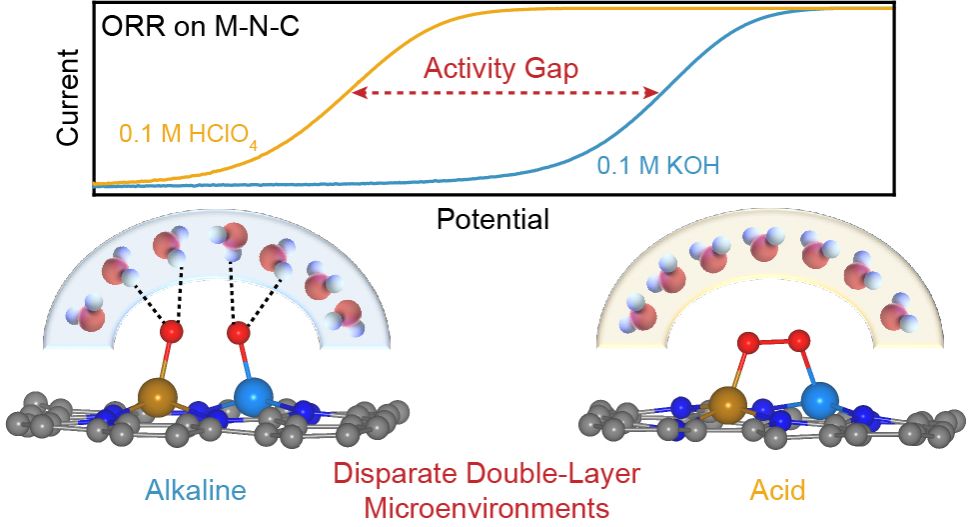
A standing puzzle in electrochemistry is that why the metal-nitrogen-carbon catalysts generally exhibit dramatic activity drop for oxygen reduction when traversing from alkaline to acid. Here, taking FeCo-N6-C double-atom catalyst as a model system and combining the ab initio molecular dynamics simulation and in situ surface-enhanced infrared absorption spectroscopy, we show that it is the significantly distinct interfacial double-layer structures, rather than the energetics of multiple reaction steps, that cause the pH-dependent oxygen reduction activity on metal-nitrogen-carbon catalysts. Specifically, the greatly disparate charge densities on electrode surfaces render different orientations of interfacial water under alkaline and acid oxygen reduction conditions, thereby affecting the formation of hydrogen bonds between the surface oxygenated intermediates and the interfacial water molecules, eventually controlling the kinetics of the proton-coupled electron transfer steps. The present findings may open new and feasible avenues for the design of advanced metal-nitrogen-carbon catalysts for proton exchange membrane fuel cells.
https://www.nature.com/articles/s41467-023-42749-7


 LINK
LINK