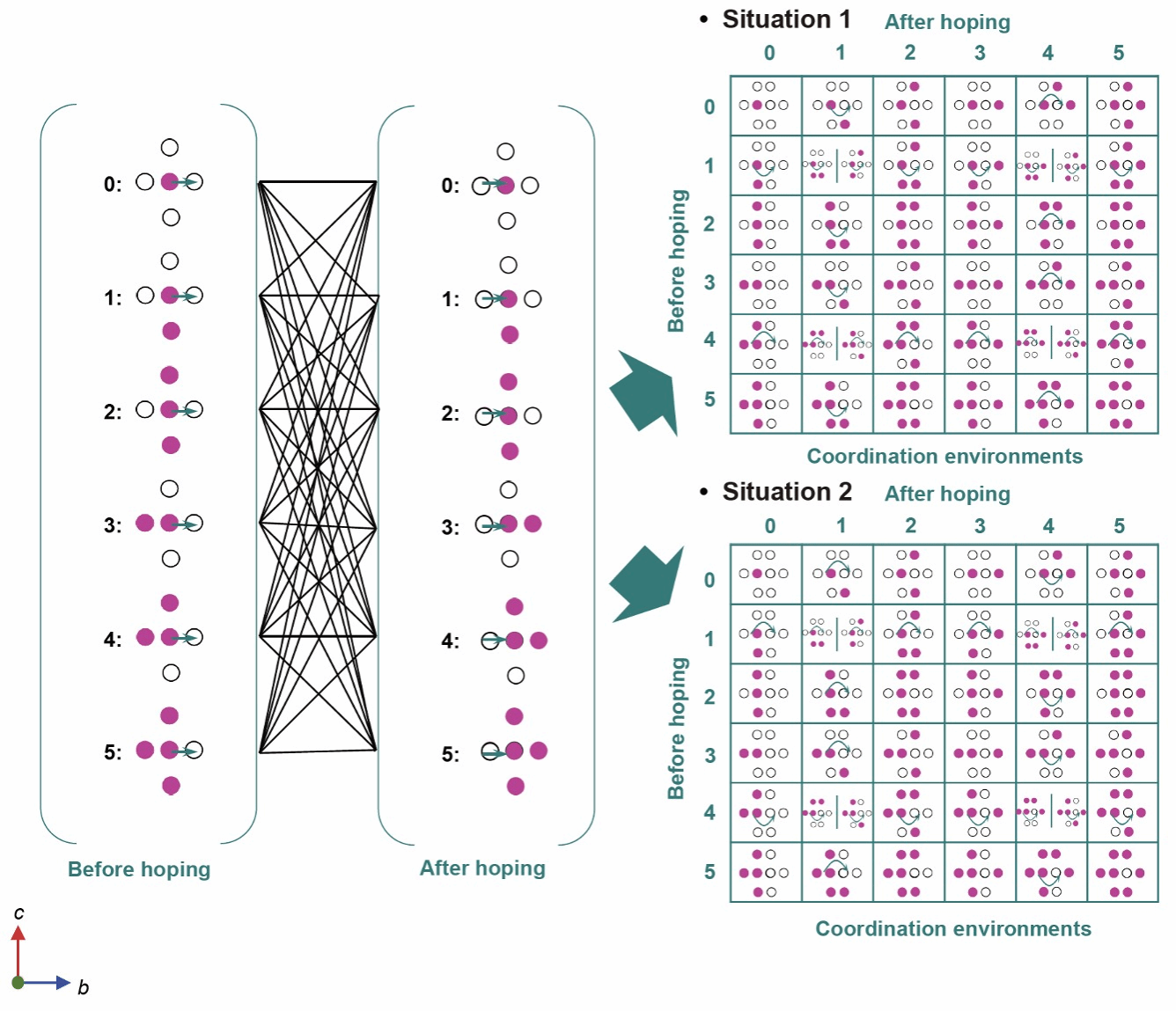
LiFePO4, one of the mainstream cathode materials of current EV batteries, exhibits experimental diffusion coefficients (Dc) of Li+ which are not only several orders of magnitude lower than those predicted by the ionic hopping barriers obtained from theoretical calculations and spectroscopic measurements, but also span several orders from 10−14 to 10−18 cm2 s−1 under different states of charge (SOC) and the charging rates (C-rates). Atomic level understanding of such sluggishness and diversity of Li+ transport kinetics would be of significance in improving the rate performance of LiFePO4 through material and operation optimization but remain challenging. Herein, we show that the high sensitivity of Li hopping barriers on the local Li–Li coordination environments (numbers and configurations) plays a key role in the ion transport kinetics. This is due a neural network-based deep potential (DP) which allows accurate and efficient calculation of hopping barriers of Li+ in LiFePO4 with various Li–Li coordination environments, with which the kinetic Monte-Carlo (KMC) method was employed to determine the Dc values at various C-rates and SOC across a broad spectrum. Especially, an accelerated KMC simulation strategy is proposed to obtain the Dc values under a wide range of SOC at low C-rates, which agree well with that obtained from the galvanostatic intermittent titration technique (GITT). The present study provides accurate descriptions of Li+ transport kinetics at both very high and low C-rates, which remains challenging to experiments and first-principles calculations, respectively. Finally, it is revealed that the gradient distributions of Li+ density along the diffusion path result in great asymmetry in the barriers of the forward and backward hopping, causing very slow diffusion of Li+ and the diverse variation of Dc.
https://doi.org/10.1007/s11426-023-1662-9


 LINK
LINK