电化学界面是电催化反应实际发生的场所,其微观结构深刻影响着界面反应的动力学与选择性。因此,理解电催化体系的表界面结构,尤其是在原子-分子水平上,并建立其与电催化反应动力学之间的联系,对于促进基础电化学学科的发展以及各种电化学技术的进步都至关重要。
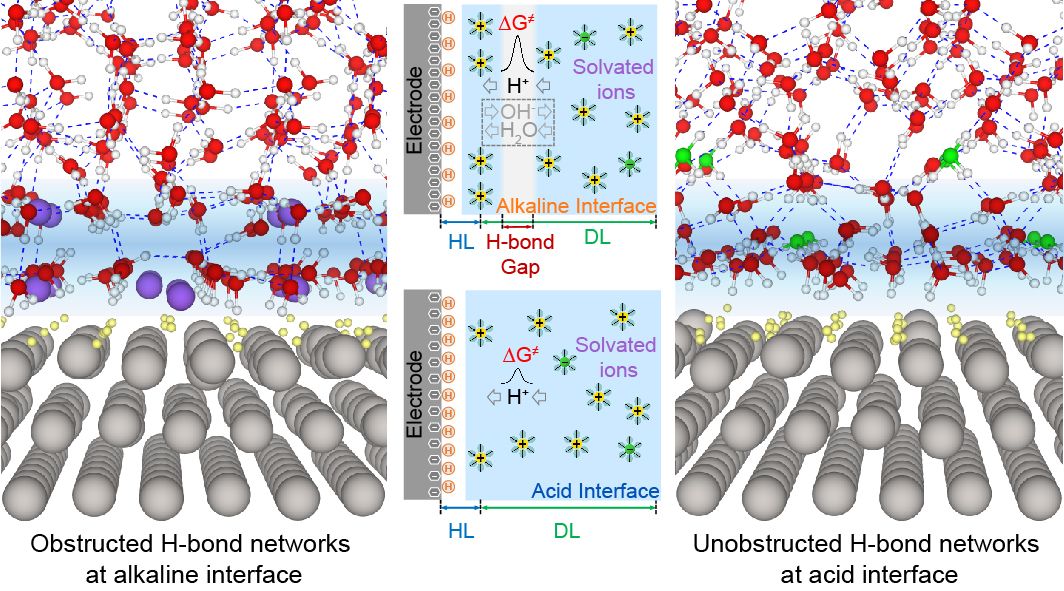
长期以来,人们关于氢电极反应动力学在碱性条件下显著减缓的原因始终难以达成共识。当前,对这一动力学pH效应的研究主要考虑*H和*OH等中间体的吸附对表面反应步骤能量的影响,而忽略了界面双电层结构对pH的依赖。因此,准确获得不同pH条件下氢电催化体系的界面双电层微观结构特征,将是解决这一难题的突破点。然而,目前在原子、分子尺度解析电催化体系界面结构及过程仍是一个巨大的挑战,任何单一技术都显不足。
为此,陈胜利教授团队结合从头算分子动力学(AIMD)模拟和原位表面增强红外光谱(in situ SEIRAS)技术,通过对AIMD模拟的酸性和碱性界面双电层结构、以及界面水分子的计算振动谱学与SEIRAS实验谱学的细致对比,提出界面双电层中氢键网络连通性的显著差异是导致氢电催化巨大动力学pH效应的根源。随后,以Pt-Ru合金为模型催化剂,进一步揭示了OHad吸附中间体在改善碱性氢电催化反应动力学的本质,即增加双电层中氢键网络的连通性,而不是如以往认为的那样,仅仅影响表面反应路径及其能量。该研究突出了界面双电层结构在电催化中的关键作用,并为结合AIMD模拟、计算谱学和实验谱学研究电化学界面原子结构提供了一个研究范式。

(通讯员:王晓晓)


 LINK
LINK